
Diseases Studied
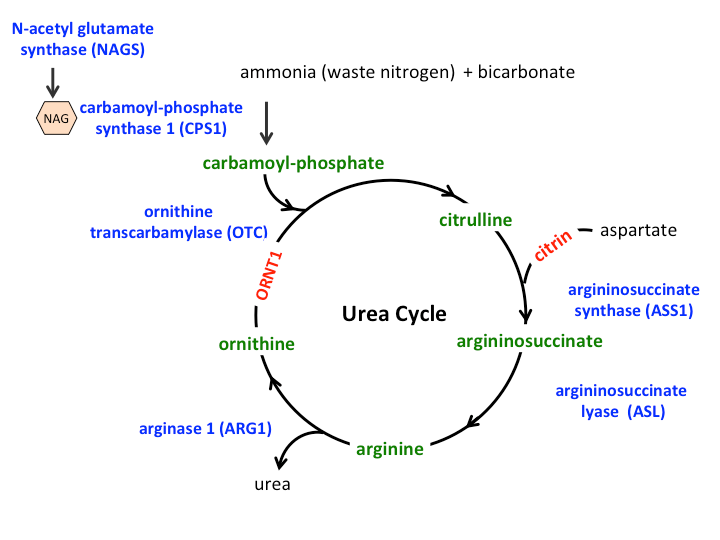
There can be an overlap in the symptoms of the different urea cycle disorders because they affect the body in the similar ways. Hence, descriptions of an individual disorder and the methods used to diagnose a particular disorder may be similar to other urea cycle disorders. The figure of the urea cycle illustrated here shows the role of the enzymes (depicted in blue) and transporters (depicted in red) in conversion of ammonia nitrogen into urea. The nitrogen from ammonia and aspartate is “handed over” to a number of intermediate compounds (depicted in green) before finally being converted into urea. A deficiency of any of the enzymes or transporters of the urea cycle leads to an inability or reduced ability to dispose of nitrogen and accumulation of ammonia.
N-acetylglutamate Synthase (NAGS) Deficiency
The enzyme NAGS makes a molecule called N-acetylglutamate, which is essential for the functioning of the first urea cycle enzyme, CPS1. Patients with complete NAGS deficiency develop high ammonia levels in the blood (hyperammonemia) soon after birth. Patients who are successfully rescued from high ammonia are at risk for further episodes of hyperammonemia. Patients with partial NAGS deficiency (milder type of NAGS) can have symptoms that appear at any time of life with triggering events such as an infection or other stress. NAGS deficiency is typically diagnosed by genetic testing. NAGS deficiency is the only UCD in which the hyperammonemia can be completely reversed by a medication called Carglumic acid.
Learn More from GARDCarbamoyl-phosphate Synthase 1 (CPS1) Deficiency
CPS1 is an enzyme that is important for the first step of the urea cycle that converts ammonia into a compound called carbamoyl-phosphate. Patients with complete CPS1 deficiency rapidly develop hyperammonemia soon after birth. Patients who are successfully rescued from hyperammonemia are at risk for repeated episodes of hyperammonemia. Patients with partial CPS1 deficiency (milder type) can have symptoms appear at any time of life with a triggering event such as an infection or other stress. CPS1 deficiency can be suspected based on biochemical tests done on blood and urine, but a definite diagnosis requires genetic testing or enzyme testing of a liver biopsy.
Ornithine Transcarbamylase (OTC) Deficiency
The enzyme OTC combines carbamyl-phosphate that is produced by CPS1 with an amino acid called ornithine to make citrulline. Patients with complete OTC deficiency (the most severe type of this disorder) rapidly develop high levels of ammonia in the blood, soon after birth and develop symptoms any time before one week of age. Infants who are successfully rescued from this first crisis are at risk for repeated episodes of hyperammonemia. The OTC gene is located on the X-chromosome. Males have only one X-chromosome while females have two. Hence, the majority of patients with severe presentations of OTC deficiency are males. Females with one “abnormal OTC gene” and one “normal OTC gene” may not show any clinical evidence of the disorder; however, 15% can show some symptoms or signs of the disease. Patients with partial OTC deficiency (milder type of OTC) can present at any time of life with a triggering event such as an infection or other stress. The rise in ammonia levels is generally lower in females with this disorder as compared to the males. OTC deficiency is typically suspected based on symptoms and biochemical tests, but a definite diagnosis requires genetic testing or enzyme testing of a liver biopsy.
Learn More from GARDArgininosuccinate Synthase (ASS) Deficiency (also known as Citrullinemia type I)
The enzyme ASS1 (or ASS) uses the citrulline produced by OTC and combines it with the amino acid aspartate to make a compound called argininosuccinate. Patients with complete deficiency of ASS (most severe type of this disorder) present with high levels of ammonia soon after birth. The blood level of citrulline in these patients is typically many times higher than the normal. The specific diagnosis can be made by plasma amino acid analysis based on extremely elevated citrulline levels and/or by enzyme analysis of cultured skin cells obtained from a skin biopsy, or by genetic testing.
Learn More from GARDCitrin Deficiency (also called Citrullinemia type II)
Citrin is a protein that is needed to transport the amino acid aspartate into the urea cycle. This type of protein is called a transporter. Adults with citrin deficiency (also called citrullinemia type II) can present with hyperammonemia, and cyclical behavior abnormalities including aggression, irritability, and hyperactivity, as well seizures, and coma. Infants and children with citrin deficiency present differently and can have abnormalities of liver and failure to thrive. These patients can also have the dietary peculiarity of avoiding sugars rather than protein (which most urea cycle patients avoid). The majority of patients reported have been Japanese or Asian who share common mutations in the Citrin gene. Citrin deficiency is typically diagnosed by a biochemical analysis and genetic testing.
Learn More from GARDArgininosuccinate Lyase (ASL) Deficiency (also known as Argininosuccinic Aciduria)
Argininosuccinate lyase is an enzyme that is needed to breakdown a compound in the urea cycle called argininosuccinate. Patients with complete deficiency of ASL (most severe type of this disorder) present soon after birth with high levels of ammonia. Those with milder forms of deficiency may present later in childhood with hyperammonemia during stress or infections. Patients can also develop trichorrhexis nodosa, a fragile hair abnormality. Patients with this condition can also develop abnormalities of the liver. Some affected patients who have never had severe hyperammonemia can also demonstrate some developmental disabilities. The levels of argininosuccinic acid in the blood and urine are high in patients (hence the name argininosuccinic aciduria). The specific diagnosis can be made by plasma amino acid analysis based on elevated citrulline and argininosuccinic acid levels and/or by enzyme analysis of cultured skin cells obtained from a skin biopsy, or by genetic testing.
Learn More from GARDArginase (ARG) Deficiency (also known as hyperargininemia)
Arginase is the last enzyme of the urea cycle that breaks down the amino acid arginine produced by the urea cycle, into two molecules, urea and ornithine. The urea is disposed of by the kidneys; this is the way nitrogen from ammonia is excreted by the body. Arginase deficiency is typically not characterized by severe increase in ammonia. Patients often present with progressive problems of muscle control. They can also develop seizures and have developmental disabilities. Growth is usually slow and without therapy they usually do not reach normal adult height. Other symptoms that may present early in life include episodes of irritability, poor appetite, and vomiting. Severe episodes of hyperammonemia can occur but are infrequent. The diagnosis of hyperargininemia is made by the elevated levels of arginine levels in the blood and by analysis of enzymatic activity in red blood cells or by genetic testing.
Learn More from GARDOrnithine Translocase Deficiency (also known as hyperornithinemia-hyperammonemia-homocitrullinuria or HHH Syndrome)
Ornithine translocase is a transport protein that moves ornithine and citrulline molecules within the urea cycle. When this transport does not work properly, it causes the urea cycle to slow down and ammonia builds up in the blood. A number of other molecules also build up including ornithine in the blood and homocitrulline in the urine. Most patients have episodic hyperammonemia, accompanied by vomiting, sleepiness and (in extreme cases) coma. Growth is abnormal and learning can be affected. Common symptoms include seizures and muscle control problems. Patients that have partial activity of the transporter (mild type of disorder) have symptoms beginning in adulthood. They typically self-select low protein diets without being aware they have this disorder. Diagnosis can be made by detecting elevated levels of plasma ornithine and urinary homocitrulline. Testing of a skin biopsy can also confirm the diagnosis.
Learn More from GARD