
Urea Cycle Treatment Guidelines
Diagnosis
The most important step in diagnosing urea cycle disorders is clinical suspicion of the presence of hyperammonemia. A plasma ammonia level is the most important laboratory test in evaluating a patient with a suspected urea cycle defect. Particular care should be taken in drawing a blood ammonia since there is significant variability depending on proper drawing technique and sample handling. Treatment should not be delayed in efforts to reach a final diagnosis; later stages of treatment should be tailored to the specific disorder. In addition to plasma ammonia, laboratory data useful in the diagnosis of UCDs include, pH, CO2, anion gap, blood lactate, plasma acylcarnitine profile acylcarnitines, plasma and urine amino acids, and urine organic acid analyses including the specific determination of orotic acid.1,2 Patients with true urea cycle defects will typically have normal glucose and electrolyte levels. The pH and CO2 can vary with the degree of cerebral edema and hyper- or hypo-ventilation but will initially reflects a respiratory alkalosis. In neonates, the normal ammonia level is higher that of adults, which typically is less than 35 µmol/L (less than 100 µmol/L in neonates). An elevated plasma ammonia level of 150 µmol/L (>260 µg/dl) or higher in neonates and > 100 µmol/l (175 µg/dl) in older children and adults, associated with a normal anion gap and a normal blood glucose level, is an indication for the presence of a urea cycle defect. Quantitative amino acid analysis can be used to evaluate these patients and arrive at a tentative diagnosis. Elevations or reduction of arginine, citrulline, and argininosuccinate (Figure 1, Diagnostic Flow Chart) will give clues to the location of defect in the cycle. The amino acid profile in sick newborns can be quite different from those in children and adults which should be taken into account.2 The levels of the nitrogen buffering amino acid glutamine can also be quite high in Ucd and can serve as an indication of an increased ammonia load. If a defect in NAGS, CPSI, or OTC is suspected, the presence or absence of elevated orotic acid in the urine can help narrow the diagnosis. Orotic acid is produced when there is an overabundance of carbamyl phosphate which spills into the pyrimidine biosynthetic system. The determination of urine organic acids and plasma acylcarnitines will also herald the presence of an organic aciduria. Other genetic defects that affect ammonia detoxification are lysinuric protein intolerance, carbonic anhydrase VA, hyperinsulinism-hyperammonemia syndrome, hypoprolinemia (paradoxical fasting hyperammonemia) and pyruvate cxarboxylase deficiency. All of these are very rare and important clues for them would be obtained through the investigations outlined above.
Figure 1. Diagnostic Flow Chart for Acute Hyperammonemia
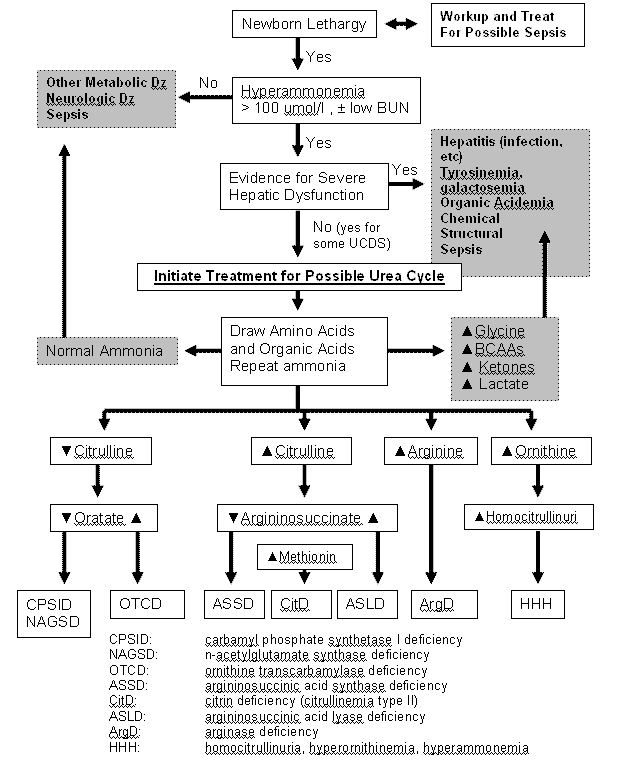
Enzymatic and DNA diagnoses is available for all of these disorders. For CPSI, OTC, and NAGS, enzymatic diagnosis can be made by DNA analysis and/or a liver biopsy specimen freshly frozen in liquid nitrogen.3 Enzymatic testing for ASS and ASL can be done on cultured skin fibroblasts and arginase can be tested on red blood cells.4 Clinically approved DNA sequence analysis is available for all the urea cycle disorders.
Treatment
Treatment of Urea Cycle Disorders
This section provides an overview of UCD management.2, 5-8 The treatment of these patients requires a highly-coordinated team of specialists trained in caring for patients with inborn errors of metabolism. Emergency management of patients in hyperammonemic coma resulting from a UCD is based on three interdependent principles: first, physical removal of the ammonia by dialysis or some form of hemofiltration; second, reversal of the catabolic state through caloric supplementation and in extreme cases, hormonal suppression (glucose/insulin drip); and third, pharmacologic scavenging of excess nitrogen. These are not consecutive but should be pursued independently in parallel as quickly as possible.8-10 (Figure 3, Emergency Management)
Figure 2: Treatment Team and Organization
- Metabolic Specialist
- coordinate treatment and management
- Intensive care team
- assist with physiologic support
- ventilator management
- sedation and pain management
- Nephrologist or dialysis team
- manage dialysis
- manage renal complications
- Surgical team
- large bore catheter placement
- liver biopsy as necessary
- gastrostomy tube placement (if indicated)
- Pharmacy staff
- formulate nitrogen scavenging drugs
- cross check dosing orders in complex management
- Laboratory staff
- analyze large number of STAT ammonia samples in the acute phase
- analyze amino acids and other specialty labs
- Nursing staff
- execute complex and rapidly changing management plan
- closely monitor patient for signs of deterioration or change
- Nutritionist
- maximize caloric intake with neutral nitrogen balance
- educate family in management of complex very low-protein diet
- Social work
- rapidly identify resources for complex outpatient treatment regimen
- work with families in highly stressful clinical situation
- Genetic Counselor
- educate family in genetics of rare metabolic disease
- identify other family members at potential risk (OTC particularly)
- ensure proper samples are obtained for future prenatal testing
- contact research/diagnostic centers for genetic testing
Figure 3: Emergency Management
- Fluids, dextrose, and interlipid to mitigate catabolism and typical dehydration (attempt 80 Kcal/kg/day)
- Septic workup and antibiotics to treat potential triggering events or primary infection
- Contact and possible transport to treatment-capable institute as soon as possible
- Remove protein from intake (P.O. or TPN)
- Establish central venous access
- Provide physiologic support (pressors, buffering agents). (Renal output is critical to long term success).
- Stabilize airway as cerebral edema may result in sudden respiratory arrest, consider prophylactic intubation and artificial ventilation.
If the patient show CNS compromise and ammonia does not fall quickly (within 2-3 hours) with high-calorie infusion plus pharmaceutical measures, central venous access should be established at once and dialysis begun immediately at the highest available flow rate. Dialysis is very effective for the removal of ammonia and the clearance is dependent on the flow through the dialysis circuit.11, 12 In severe cases of hyperammonemia, provision for hemofiltration should be made to follow the dialysis until the patient is stabilized and the catabolic state is reversed. Some patients will show rebound hyperammonemia after their initial round of dialysis and may require additional periods of dialysis. Typically, continuous hemofiltration can successfully mitigate this rebound.
The importance of the management of the catabolic state cannot be overstressed.6 Since the catabolism of protein stores is often the triggering event for hyperammonemia, the patient will not stabilize and ammonia will continue to rise until it is reversed. Fluids, dextrose and Intralipid® should be given to help blunt the catabolic process, however, catabolism will not fully reverse without the supplementation of amino acids or protein. This should start as soon as possible, usually once the ammonia level can be maintained under 200umol/l. The patient should be assessed for dehydration and fluids replaced. Since these patients suffer from cerebral edema, care should be taken to avoid over hydration. The nitrogen scavenging drugs are usually administered in a large volume of fluid which should be taken into consideration. A regimen of 80-120 kcal/kg/day is a reasonable goal. The administration of insulin and glucose can be useful but care should be exercised, to avoid rapid swings in blood glucose concentrations to prevent rapid osmotic shifts in the brain. Supplementation of arginine (except in argininemia) serves to replace arginine not produced by the urea cycle enzyme (in addition to the partial cycle function it can stimulate) and prevents its deficiency from causing additional protein catabolism. Refeeding the patient as soon as practicable is useful since more calories can be administered this way. The use of essential amino acid formulations in feeding can reduce the amount of protein necessary to meet basic needs.6 Table 1 is reproduced from Singh et al and lists the proposed caloric needs for patients with urea cycle disorders.6
Table 1: Recommended Daily Nutrient Intakes (Ranges) for Infants, Children, and Adults With Urea Cycle Disorders
| Nutrient | ||||
|---|---|---|---|---|
| Age | Protein | Patient Kcal Intake | Energy | Fluid |
| Infants | ||||
| Girls and Boys | ||||
| Women | ||||
| Men | ||||
| 0 to 3 months | 2.20 - 1.25(g/kg) | 150 - 101(kcal/kg) | 150 - 125(kcal/kg) | 160 - 130(mL/kg) |
| 3 to 6 months | 2.00 - 1.15 (g/kg) | 100 - 80 (kcal/kg) | 140 - 120 (kcal/kg) | 160 - 130 (mL/kg) |
| 9 to 12 months | 1.60 - 0.90 (g/kg) | 80 - 75 (kcal/kg) | 120 - 110(kcal/kg) | 130 - 120(mL/kg) |
| 1 to 4 years | 8 - 12 (g/day) | 800 - 1040 (kcal/day) | 945 - 1890 (kcal/day) | 945 - 1890 (mL/day) |
| 4 to 7 years | 12 - 15 (g/day) | 1196 - 1435 (kcal/day) | 1365 - 2415 (kcal/day) | 1365 - 2245 (mL/day) |
| 7 to 11 years | 14 - 17 (g/day) | 1199 - 1693 (kcal/day) | 1730 - 3465 (kcal/day) | 1730 - 3465 (mL/day) |
| 11 to 15 years | 20 - 23 (g/day) | 1575 - 3150 (kcal/day) | 1575 - 3150 (mL/day) | |
| 15 to 19 years | 20 - 23 (g/day) | 1260 - 3150 (kcal/day) | 1260 - 3150 (mL/day) | |
| less than or equal to 19 years | 22 - 25 (g/day) | 1785 - 2625 (kcal/day) | 1875 - 2625 (mL/day) | |
| 11 to 15 years | 20 - 23 (g/day) | 2100 - 3885 (kcal/day) | 2100 - 3885 (mL/day) | |
| 15 to 19 years | 21 - 24 (g/day) | 2200 - 4095 (kcal/day) | 2200 - 4095 (mL/day) | |
| less than or equal to 19 years | 23 - 32 (g/day) | 2625 - 3465 (kcal/day) | 2625 - 3465 (mL/day) | |
Emergency pharmacologic management with ammonia scavengers and L-arginine is initiated as soon as possible using the drug combination sodium phenylacetate and sodium benzoate (Ammonul), ideally while the dialysis is being arranged and the diagnostic workup is under way. Two agents are used in combination to trap nitrogen in excretable forms. Sodium benzoate combines with glycine to make hippurate which is excreted by the kidneys (or removed in the dialysate), and sodium phenylacetate combines with glutamine to make phenacetylglutamine which is also excreted in the urine.13,14 The body replaces these amino acids using excess nitrogen. It has proposed that the removal of glutamine by phenylacetate has the additional benefit of removing a compound suspected of having a major role in the neurotoxicity of these disorders15-20 but it is unclear if plasma glutamine has any effect on the brain. The administration of a second loading dose of these drugs to the patient after the initial phase is not recommended due to concerns of toxicity. L-arginine must also be administered continuously in the acute phase of treatment of urea cycle disorders. In addition to replenishing circulating amino acid levels, arginine can utilize those parts of the cycle not affected by the genetic blocks and incorporate some nitrogen into urea cycle amino acids citrulline and argininosuccinate. Since arginine is the precursor for nitric oxide production, it is worth considering modification of the arginine dose downward if the patient develops vasodilation and hypotension. Table 2 lists drug doses for the acute management by diagnosis (information extracted from FDA package insert). Due to the potential for toxicity (lethal in extreme cases) of these drugs consultation with an experienced metabolic physician is recommended before starting treatment.21 A resource for finding these physicians and other treatment suggestions is found in the home page for this website.
Table 2: Ammonul Dosage and Administration Summary Tables
Patient Population: Neonates to Young Children: NAGS, CPS and OTC Deficiency
| Components of Infusion Solution | Dosage Provided | |||||
|---|---|---|---|---|---|---|
| Ammonul | Arginine HCl Injection, 10% | Dextrose Injection, 10% | Sodium Phenylacetate | Sodium Benzoate | Arginine HCl | |
| Loading Dose (90 min) | 2.5 mL/kg | 2.0 mL/kg | greater or equal to 25 mL/kg | 250 mg/kg | 250 mg/kg | 200 mg/kg |
| Maintenance Dose | 2.5 mL/kg/24 hr | 2.0 mL/kg/24 hr | greater or equal to 25 mL/kg | 250 mg/kg/24 hr | 250 mg/kg/24 hr | 200 mg/kg/24 hr |
Patient Population: Neonates to Young Children: Unknown, ASD and ASL Deficiency
| Components of Infusion Solution | Dosage Provided | |||||
|---|---|---|---|---|---|---|
| Ammonul | Arginine HCl Injection, 10% | Dextrose Injection, 10% | Sodium Phenylacetate | Sodium Benzoate | Arginine HCl | |
| Loading Dose (90 min) | 2.5 mL/kg | 6.0 mL/kg | greater or equal to 25 mL/kg | 250 mg/kg | 250 mg/kg | 600 mg/kg |
| Maintenance Dose | 2.5 mL/kg/24 hr | 6.0 mL/kg/24 hr | greater or equal to 25 mL/kg | 250 mg/kg/24 hr | 250 mg/kg/24 hr | 600 mg/kg/24 hr |
Patient Population: Older Children and Adults: NAGS, CPS and OTC Deficiency
| Components of Infusion Solution | Dosage Provided | |||||
|---|---|---|---|---|---|---|
| Ammonul | Arginine HCl Injection, 10% | Dextrose Injection, 10% | Sodium Phenylacetate | Sodium Benzoate | Arginine HCl | |
| Loading Dose (90 min) | 55 mL/m 2 | 2.0 mL/kg | greater or equal to 25 mL/kg | 5.5 g/m 2 | 5.5 g/m 2 | 200 mg/kg |
| Maintenance Dose | 55 mL/m 2 /24 hr | 2.0 mL/kg/24 hr | greater or equal to 25 mL/kg | 5.5 g/m 2 /24 hr | 5.5 g/m 2 /24 hr | 200 mg/kg/24 hr |
Patient Population: Unknown, ASD and ASL Deficiency
| Components of Infusion Solution | Dosage Provided | |||||
|---|---|---|---|---|---|---|
| Ammonul | Arginine HCl Injection, 10% | Dextrose Injection, 10% | Sodium Phenylacetate | Sodium Benzoate | Arginine HCl | |
| Loading Dose (90 min) | 55 mL/m 2 | 6.0 mL/kg | greater or equal to 25 mL/kg | 5.5 g/m 2 | 5.5 g/m 2 | 600 mg/kg |
| Maintenance Dose | 55 mL/m 2 /24 hr | 6.0 mL/kg/24 hr | greater or equal to 25 mL/kg | 5.5 g/m 2 /24 hr | 5.5 g/m 2 /24 hr | 600 mg/kg/24 hr |
After the initial loading phase and dialysis, the patient's dose should be converted to the maintenance doses of the ammonia scavengers listed in the manufacturers packaging insert (Table 1). If the exact enzyme defect is known the amount of arginine administered can be adjusted downward. If chronic therapy is warranted the patient can then be switched to the oral pro-drug of phenylacetate, phenylbutyrate (Buphenyl). The usual total daily dose of Buphenyl Tablets and Powder for patients with urea cycle disorders is 450 - 600 mg/kg/day in patients weighing less than 20 kg, or 9.9 - 13.0g/m2/day in larger patients. The tablets and powder are to be taken in equally divided amounts with each meal or feeding (i.e., three to six times per day). Citrulline supplementation is recommended for patients diagnosed with deficiency of n-acetylglutamate synthase, carbamylphosphate synthetase or ornithine transcarbamylase; citrulline; daily recommended intake is 0.17g/kg/day or 3.8g/m2/day. Arginine supplementation is needed for patients diagnosed with deficiency of argininosuccinic acid synthetase; arginine (free base) daily intake is recommended at 0.4 - 0.7g/kg/day or 8.8 - 15.4g/m2/day. In patients with n-acetylglutamate synthetase, the use of carbamyl glutamate has been demonstrated to be very effective22, and there is a study investigating its utility.
Treatment issues
Other Treatment Issues
During acute severe hyperammonemia, ventilator or circulatory support may be required Anticonvulsive medication to control seizures and sedation or head cooling to reduce cerebral activity could be of benefit to these patients but has not been clinically evaluated. Any infection should be treated vigorously. Electrolytes and acid-base balance should to be checked every 6 hours during the initial phase of treatment. The use of osmotic agents such as mannitol is not felt to be effective in treating the cerebral edema from hyperammonemia but this is mainly anecdotal. In canines, opening the blood brain barrier with mannitol resulted in cerebral edema by promoting the entry of ammonia into the brain fluid compartment.23, 24 Administration of steroids and valproic acid should be avoided. Other measures include physiologic support (pressors, buffering agents to maintain pH and buffer arginine HCl) and maintenance of renal output, particularly if ammonia scavengers are being used. Finally, it is imperative to reassess continuation of care after the initial phase of treatment.
Rapid response to reduce the hyperammonemia is indispensable for a good outcome.25, 26 The pathophysiology centers around cerebral edema, disruptions in neurochemistry and pressure on the brainstem. The resulting decrease in cerebral blood flow plus prolonged seizures, when they occur, are poor prognostic factors. In adults, because the sutures of the skull are fused, sensitivity to hyperammonemia appears considerably greater than in infants.8 Thus treatment should be aggressive and intensified at a lower ammonia concentration than in children.
Neurologic Evaluation: Cerebral studies should be conducted to determine the efficacy of treatment and whether continuation is warranted. EEG should be performed to assess both cerebral function and evidence of seizure activity. If available, MRI determined cerebral blood flow can be used to establish if venous stasis has occurred from cerebral edema. Evaluation of brain stem function and higher cortical function are useful to assess outcome. Finally, in the most severe cases, the decision for continuation of critical care support takes into consideration the neurologic status, duration of the patient's coma and potential for recovery, and whether the patient can be a candidate for transplantation among others including ethical considerations. If the basic urea cycle defect is severe enough, liver transplantation should be considered. Criteria for transplantation are of course linked back to neurologic status, duration of coma, and availability of donor organs. Diagnostic samples of DNA, liver, and skin should be obtained since they can be important in family counseling and future treatment issues.
Long Term Management
Every effort should be made to avoid triggering hyperammonemia events. It is imperative to prevent or quickly interrupt a catabolic state at an early stage of impending decompensation during subsequent illnesses or surgeries, as well as during any event resulting in significant bleeding or tissue damage. As this usually happens at home, it is essential to educate the family about how to react adequately.7, 8, 27, 28 Patients should carry an emergency card or bracelet containing essential information and phone numbers as well as instructions on emergency measures. Every patient should relate to physicians and a hospital with a dedicated team of metabolic specialists who can be reached at any time. For vacations it is usually prudent to inquire about metabolic services in the respective destination.
Long-term dietary modification with nutritional oversight is often necessary in patients with UCD. Patients should also avoid dehydration, an especially common occurrence among adults in connection with alcohol intake, hiking, and airline flights. Not all adult patients who recover from a hyperammonemic episode require chronic nitrogen scavengers, but they ought to be considered since many of these patients can become more brittle as time goes on. In particular, steroids and valproic acid administration are contraindicated since they can precipitate hyperammonemia.
Should psychiatric problems occur over the long term, caregivers should be alert to the possibility of hyperammonemia. In addition, patients with citrullinemia type II, in particular, have presented with mental disturbances.29
Clinical observations of patients with ASL deficiency demonstrate a high incidence of chronic progressive fibrosis of the liver. This finding is can be seen, but much less frequently in other urea cycle disorders and studies are underway to better determine the exact pathophysiology. It is important to provide genetic counseling in order to assess risk to other family members.
Reference List
- Steiner RD, Cederbaum SD. Laboratory evaluation of urea cycle disorders. J Pediatr. 2001; 138(1 Suppl):S21-S29.
- Summar M. Current strategies for the management of neonatal urea cycle disorders. J Pediatr. 2001; 138(1 Suppl):S30-S39.
- Tuchman M, Tsai MY, Holzknecht RA, Brusilow SW. Carbamyl phosphate synthetase and ornithine transcarbamylase activities in enzyme-deficient human liver measured by radiochromatography and correlated with outcome. Pediatr Res. 1989; 26(1):77-82.
- Steiner RD, Cederbaum SD. Laboratory evaluation of urea cycle disorders. J Pediatr. 2001; 138(1 Suppl):S21-S29.
- Summar ML. Presentation and management of urea cycle disorders outside the newborn period. Crit Care Clin. 2005; 21(4 Suppl):ix.
- Singh RH, Rhead WJ, Smith W, Lee B, King LS, Summar M. Nutritional management of urea cycle disorders. Crit Care Clin. 2005; 21(4 Suppl):S27-S35.
- Lee B, Singh RH, Rhead WJ, Sniderman KL, Smith W, Summar ML. Considerations in the difficult-to-manage urea cycle disorder patient. Crit Care Clin. 2005; 21(4 Suppl):S19-S25.
- Summar ML, Barr F, Dawling S et al. Unmasked adult-onset urea cycle disorders in the critical care setting. Crit Care Clin. 2005; 21(4 Suppl):S1-S8.
- Summar M. Current strategies for the management of neonatal urea cycle disorders. J Pediatr 2001; 138(1 Suppl):S30-S39.
- Summar M, Pietsch J, Deshpande J, Schulman G. Effective hemodialysis and hemofiltration driven by an extracorporeal membrane oxygenation pump in infants with hyperammonemia. J Pediatr. 1996; 128(3):379-382.
- Summar M. Current strategies for the management of neonatal urea cycle disorders. J Pediatr. 2001; 138(1 Suppl):S30-S39.
- Summar M, Pietsch J, Deshpande J, Schulman G. Effective hemodialysis and hemofiltration driven by an extracorporeal membrane oxygenation pump in infants with hyperammonemia. J Pediatr. 1996; 128(3):379-382.
- Batshaw ML. Sodium benzoate and arginine: alternative pathway therapy in inborn errors of urea synthesis. Progress in Clinical & Biological Research. 1983; 127:69-83.
- Batshaw ML, Brusilow SW. Evidence of lack of toxicity of sodium phenylacetate and sodium benzoate in treating urea cycle enzymopathies. Journal of Inherited Metabolic Disease. 1981; 4(4):231.
- Batshaw ML, Brusilow SW. Treatment of hyperammonemic coma caused by inborn errors of urea synthesis. Journal of Pediatrics. 1980; 97(6):893-900.
- Brusilow SW, Valle DL, Batshaw M. New pathways of nitrogen excretion in inborn errors of urea synthesis. Lancet. 1979; 2(8140):452-454.
- Brusilow SW. Phenylacetylglutamine may replace urea as a vehicle for waste nitrogen excretion. Pediatric Research. 1991; 29(2):147-150.
- Butterworth RF. Effects of hyperammonaemia on brain function. [Review] [75 refs]. Journal of Inherited Metabolic Disease. 1998; 21 Suppl 1:6-20.
- Connelly A, Cross JH, Gadian DG, Hunter JV, Kirkham FJ, Leonard JV. Magnetic resonance spectroscopy shows increased brain glutamine in ornithine carbamoyl transferase deficiency. Pediatric Research. 1993; 33(1):77-81.
- Willard-Mack CL, Koehler RC, Hirata T et al. Inhibition of glutamine synthetase reduces ammonia-induced astrocyte swelling in rat. Neuroscience. 1996; 71(2):589-599.
- Batshaw ML, MacArthur RB, Tuchman M. Alternative pathway therapy for urea cycle disorders: twenty years later. J Pediatr. 2001; 138(1 Suppl):S46-S54.
- Morizono H, Caldovic L, Shi D, Tuchman M. Mammalian N-acetylglutamate synthase. Mol Genet Metab. 2004; 81 Suppl 1:S4-11.
- Fujiwara M. Role of ammonia in the pathogenesis of brain edema. Acta Medica Okayama. 1986; 40(6):313-320.
- Fujiwara M, Watanabe A, Shiota T et al. Hyperammonemia-induced cytotoxic brain edema under osmotic opening of blood-brain barrier in dogs. Res Exp Med (Berl). 1985; 185(6):425-427.
- Brusilow SW. Urea cycle disorders: clinical paradigm of hyperammonemic encephalopathy. Prog Liver Dis. 1995; 13:293-309.
- Brusilow SW. Disorders of the urea cycle. Hospital Practice (Office Edition). 1985; 20:65-72.
- Smith W, Kishnani PS, Lee B et al. Urea cycle disorders: clinical presentation outside the newborn period. Crit Care Clin. 2005; 21(4 Suppl):S9-17.
- Dixon MA, Leonard JV. Intercurrent illness in inborn errors of intermediary metabolism. Arch Dis Child. 1992; 67(11):1387-1391.
- Saheki T, Kobayashi K, Iijima M et al. Adult-onset type II citrullinemia and idiopathic neonatal hepatitis caused by citrin deficiency: involvement of the aspartate glutamate carrier for urea synthesis and maintenance of the urea cycle. Mol Genet Metab. 2004; 81 Suppl 1:S20-S26.
Other Useful Resources
- Review of Urea Cycle Disorders
- Proceedings of a Consensus Conference for the Management of Patients with Urea Cycle Disorders
- Current strategies for the management of neonatal urea cycle disorders
- Gene Tests- A resource to search for molecular and clinical laboratories conducting testing to diagnose urea cycle disorders.
- NCBI Genetic Testing Registry- The Genetic Testing Registry (GTR®) provides a central location for voluntary submission of genetic testing information by providers. The scope includes the test's purpose, methodology, validity, evidence of the test's usefulness, and laboratory contacts and credentials. The overarching goal of the GTR® is to advance the public health and research into the genetic basis of health and disease.
Links